Si ringrazia l’Ing. Laura Monti, Subject Matter Expert di Adeodata srl, per il materiale fornito.
Nella prima parte del nostro approfondimento abbiamo analizzato i requisiti per la convalida dei sistemi computerizzati per medical device presenti nel:
- 21 CFR parte 820
- 21 CFR parte 11
- ISO 13485:2016
e abbiamo compreso che se il software è usato per la produzione o il sistema qualità di un medical device è necessaria la convalida del sistema per assicurare che il sistema soddisfi il suo intended use.
Se il sistema memorizza dati elettronici serve anche la conformità al 21CFR part 11.
Nella parte 2 del nostro approfondimento prenderemo in esame le linee guida per la convalida:
- ISO/TR 80002-2:2017 Medical device software – Part 2: Validation of software for medical device quality systems
- FDA General Principles of Software Validation; Final Guidance for Industry and FDA Staff, 2002
*Questo approfondimento riguarda:
- software utilizzati per la produzione di un dispositivo o per i monitoraggi e le misurazioni
- software utilizzati per la gestione del sistema qualità del produttore
FDA GENERAL PRINCIPLES OF SOFTWARE VALIDATION
La linea guida FDA, nel capitolo 6 Validation of automated process equipment and quality system software si concentra sulla verifica dalla corretta operatività del software nel contesto e per l’intended use previsto dal produttore del dispositivo medico.
Lo sforzo di convalida deve essere proporzionato al rischio legato al processo e alla complessità del software (come previsto anche dal 21CFR part 820 e dalla ISO 13485:2016).
L’esecuzione della convalida del software può essere delegata al fornitore del software/equipment o a terze parti ma il produttore del device rimane il responsabile ultimo che il software:
- sia validato in accordo a procedure scritte per il suo intended use
- operi come atteso all’interno del processo.
In questo contesto la documentazione di convalida deve includere:
- User Requirements
- Protocolli di convalida
- Evidenze dei test eseguiti
- Validation Summary che confermi esplicitamente che il software è convalidato per il suo intended use.
Gli user requirements definiscono l’intended use per il software e quanto il produttore dipende dal software la produzione di un medical device di qualità.
I protocolli di convalida devono includere test (con criteri di accettazione predeterminati) per la verifica di:
- Allarmi e condizioni di errore
- Accensione e spegnimento
- Funzioni e controlli eseguiti dall’utente
- Valori di minimo e massimo dei range
- Condizioni di stress
Anche i software OTS (off-the-shelf) – software disponibili sul mercato per l’acquisto da parte di aziende interessate all’utilizzo – devono essere verificati per l’intended use con audit al fornitore.
ISO/TR 80002-2
La ISO/TR 80002-2 è la linea guida utilizzata per rispondere al requisito di convalida software presente nella ISO 13485:2016.
I concetti chiave della ISO/TR 80002-2 sono:
- utilizzo di toolbox per la convalida
- critical thinking
- risk based approach
Quando convalidare un software?
Un software è da convalidare solo se ha impatto sulla qualità del medical device e/o se automatizza o esegue un attività richiesta dai requisiti regolatori.
Per la convalida, la ISO/TR 80002-2 propone un ciclo di vita waterfall.
E’ possibile utilizzare altri modelli di convalida purchè rispondano ai concetti chiave elencati prima (toolbox, critical thinking, risk based approach).
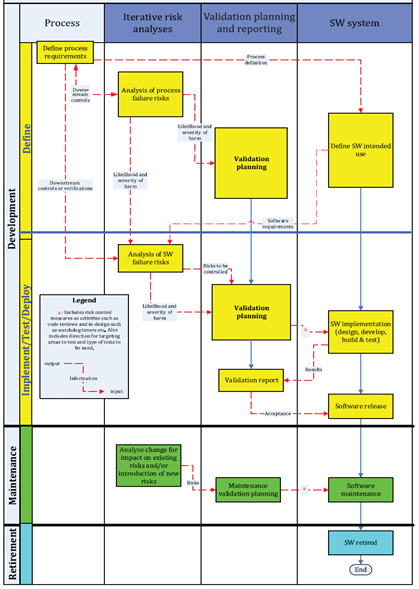
Ciclo di vita waterfall
DEFINE
Obiettivo della prima fase è definire l’intended use del software a partire dall’analisi del processo nel quale si inserisce.
Per prima cosa è necessario definire i requisiti di processo, inclusi i requisiti regolatori, tecnologici e di interfaccia.
Successivamente si esegue un’analisi documentata dei rischi di processo e delle misure di controllo del rischio concentrandosi sulla relazione tra software e rischi.
In funzione dei risultati dell’analisi dei rischi, si definisce il livello della convalida e della documentazione e si scelgono i toolbox per implementazione/test/rilascio. La scelta deve essere motivata per dimostrare che il software funzioni come atteso (Validation Planning).
In ultimo si definisce l’intended use del software all’interno del processo.
IMPLEMENT/TEST/DEPLOY
L’obiettivo di questa fase è finalizzare il Validation Plan, quindi progettare, configurare, implementare il software e testarlo.
Per prima cosa è necessario effettuare un’analisi dei rischi del software identificando le misure per ridurre l’impatto di una failure, quindi minimizzare lo sforzo e formalizzare la convalida e i controlli a valle.
Una volta finalizzato il Validation Plan, si passa all’implementazione del software grazie all’utilizzo di tool specifici per ciascuna fase di:
- implementation
- testing
- deploy.
Fondamentale è la redazione del Validation Report con documenti, attività svolte e risultati. Il Validation Report deve riportare chiaramente le conclusioni del processo di convalida.
In ultimo si formalizza il rilascio della versione convalidata del software.
MAINTENANCE
L’obiettivo della fase di maintenance è assicurare che il software rimanga in stato di convalida dopo il suo rilascio.
Allo scopo si possono utilizzare tool quali:
- backup e restore
- piani di manutenzione
- system monitoring
- test di compatibilità
- analisi dei problemi noti
- …
RETIREMENT
Ogni software prima o poi va incontro al decommissioning (attività di dismissione del sistema) o per obsolescenza o per cambiamenti di sistemi e processi.
Scopo della fase di retirement è documentare la dismissione del software e stabilire un metodo per accedere ai record per tutto il periodo di ritenzione previsto.
GAMP 5 VS ISO/TR 80002-2
Rispetto alla ISO/TR 80002-2, le GAMP 5 approcciano la convalida dal punto di vista del software (e non del processo) utilizzando un ciclo a V invece che waterfall.
Nelle GAMP 5, la strategia di convalida iniziale è basata sulla categoria del sotware indipendentemente dal contesto in cui viene utilizzato.
Per questo motivo il set documentale dipende dalla categoria del software.
L’approccio GAMP5 si applica meglio al settore farmaceutico che al settore medical device.
In ogni caso il suggerimento è di scegliere uno solo di questi due approcci e seguirlo dall’inizio alla fine del processo di convalida.
Articolo a cura di Giulia Colombo, Communication & Events Specialist di Quality Systems
Scopri i nostri servizi di convalida per i Medical Device
Leggi altre GMP NEWS di Quality Systems


