Si ringrazia l’Ing. Laura Monti, Subject Matter Expert di Adeodata srl, per il materiale fornito.
Poichè per medical device si intende qualunque strumento, apparecchio, apparecchiatura, software, impianto, reagente, materiale destinato dal fabbricante a essere impiegato sull’uomo e che non esercita sul corpo umano l’azione principale cui è destinato mediante mezzi farmacologici, immunologici o metabolici, all’interno della categoria si classificano svariati dispositivi. Per questo il quadro normativo è spesso poco specifico, in modo da adattarsi alle numerose tipologie di dispositivi presenti sul mercato.
In ambito MD si utilizzano quattro tipi di software:
- come componenti, parti o accessori di MD
- software che sono medical device
- utilizzati per la produzione di un dispositivo o per i monitoraggi e le misurazioni
- utilizzati per la gestione del sistema qualità del produttore
In questo articolo ci concentreremo solamente sulle ultime due categorie di software.
Iniziamo a vedere qual è il quadro normativo a cui bisogna fare riferimento quando si parla di convalida di software utilizzati per la produzione/monitoraggio/misurazione di un MD o per la gestione del sistema qualità.
IL QUADRO NORMATIVO
- FDA, 21 CFR part 820 Medical Devices – Quality System Regulation
- FDA, 21 CFR part 11 Electronic records and electronic signatures
- Regulation (EU) 2017/745
- Regulation (EU) 2017/746
- ISO 13485:2016 Medical devices – Quality management systems – Requirements for regulatory purposes
Linee guida per la convalida:
- ISO/TR 80002-2:2017 Medical device software – Part 2: Validation of software for medical device quality systems
- FDA General Principles of Software Validation; Final Guidance for Industry and FDA Staff, 2002
I REQUISITI DEL 21 CFR PART 820 E ISO 13485:2016
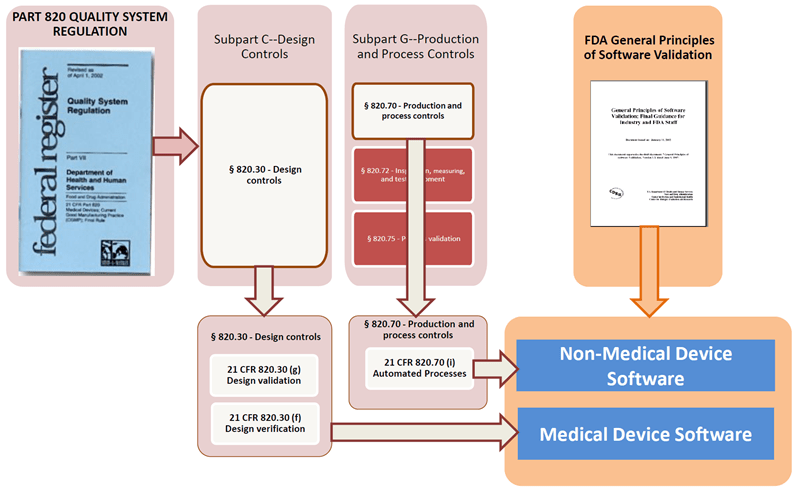
Sia nel 21CFR part 820 che nella ISO 13485 è prevista la convalida dei sistemi computerizzati. Tale convalida deve tenere conto dell’intended use del sistema e deve essere documentata (21 CFR 820, 3(z); 70 (i)) (ISO 13485 4.1.6, 7.5.6, 7.6).
Tra i requisiti di convalida troviamo specifico riferimento alla riconvalida dopo modifica (change control) (21 CFR 820, 70 (i)) (ISO 13485 4.1.6, 7.5.6, 7.6).
Lo sforzo della convalida/riconvalida deve essere proporzionato al rischio associato all’utilizzo del software (ISO 13485 4.1.6, 7.5.6, 7.6).
I requisiti di convalida riguardano anche gli equipment (21 CFR 820,70(g),72(a)) (ISO 13485 6.3, 7.5.1,7.6) che devono essere gestiti secondo procedure di taratura, controllo, manutenzione e gestione (21 CFR 820 72) (ISO 13485 6.3, 7.6).
Per quanto riguarda la conservazione dei dati, nel 21 CFR Part 820 e nella ISO 13485 è richiesta l’accessibilità ai record originali per tutto il periodo di conservazione in modo da minimizzarne il deterioramento e prevenirne la perdita (21 CFR 820 180) (ISO 13485 4.2.5).
I record archiviati in un sistema computerizzato devono inoltre essere soggetti a back-up (21 CFR 820 180).
La ISO 13485 inserisce l’audit trail tra i requisiti di un sistema computerizzato (ISO 13485 4.2.5).
In entrambe i documenti ci sono specifici riferimenti all’addestramento del personale.
I training devono essere effettuati in accordo a una procedura operativa e devono essere documentati (21 CFR 820 25) (ISO 13485 6.2).
(a) General. Each manufacturer shall have sufficient personnel with the necessary education, background, training, and experience to assure that all activities required by this part are correctly performed.
(b) Training. Each manufacturer shall establish procedures for identifying training needs and ensure that all personnel are trained to adequately perform their assigned responsibilities. Training shall be documented.
(1) As part of their training, personnel shall be made aware of device defects which may occur from the improper performance of their specific jobs.
(2) Personnel who perform verification and validation activities shall be made aware of defects and errors that may be encountered as part of their job functions.
21 CFR part 820 §25
Di fondamentale importanza risulta essere la valutazione dei fornitori sulla base della loro capacità di soddisfare i requisiti attesi, inclusi i requisiti di qualità (21 CFR 820 50) (ISO 13485 7.4.1).
Each manufacturer shall establish and maintain procedures to ensure that all purchased or otherwise received product and services conform to specified requirements.
(a) Evaluation of suppliers, contractors, and consultants. Each manufacturer shall establish and maintain the requirements, including quality requirements, that must be met by suppliers, contractors, and consultants. Each manufacturer shall:
(1) Evaluate and select potential suppliers, contractors, and consultants on the basis of their ability to meet specified requirements, including quality requirements. The evaluation shall be documented.
(2) Define the type and extent of control to be exercised over the product, services, suppliers, contractors, and consultants, based on the evaluation results.
(3) Establish and maintain records of acceptable suppliers, contractors, and consultants.
21 CFR part 820 §50
Un ulteriore requisito riguarda la gestione della documentazione. I documenti devono essere:
- rivisti e approvati, con data e firma, prima dell’emissione
- disponibili ove necessario
- ritirati quando obsoleti
- gestiti secondo procedure.
Devono inoltre esserci procedure di revisione e controllo dei change documentali (21 CFR 820 40) (ISO 13485 4.2.4) e per la corretta gestione delle non conformità e delle CAPA (21 CFR 820 100) (ISO 13485 8.5.2, 8.5.3).
L’ultimo requisito riguarda il rilascio del lotto: il sistema deve chiaramente identificare e registrare l’identità della persona che rilascia o certifica il lotto (ISO 13485 8.2.6).
I REQUISITI DEL 21 CFR PARTE 11
I requisiti presenti nel 21 CFR parte 11 sono in parte sovrapponibili a quanto previsto dal 21 CFR Parte 820 e dalla ISO 13485. Quindi è richiesta:
- la convalida dei sistemi computerizzati (§11.10 (a))
- la pronta disponibilità dei record ai fini ispettivi (§11.1 (e))
- la protezione dei record durante il periodo di conservazione (§11.10 (c))
- la verifica dell’autorità di chi esegue le operazioni (§11.10 (g))
- l’uso di audit trail generato in automatico dal sistema che:
- registri data e ora delle azioni di creazione, modifica o cancellazione di un record elettronico;
- associ l’azione a chi l’ha eseguita;
- riporti il valore prima e dopo la modifica
- un appropriato controllo della documentazione (§11.10 (k))
- l’addestramento del personale (§11.10 (i)).
A questi requisiti si aggiunge:
- la possibilità di generare copie complete ed accurate dei record per ispezioni (§11.10 (b))
- la limitazione degli accessi ai soli autorizzati (§11.10 (d))
- la verifica delle operazioni da parte del sistema, in modo da garantire la corretta sequenza delle azioni (control check) (§11.10 (f))
- la verifica che un dispositivo sia corretto per la specifica operazione (device check) (§11.10 (h))
Per i sistemi aperti – un sistema le cui componenti non sono tutte interne alla LAN aziendale – deve essere assicurata la confidenzialità, integrità e autenticità dei dati (ad esempio con la crittografia o la firma digitale) (§11.30).
LA FIRMA E I RECORD ELETTRONICI
Proprio sulla firma e i record elettronici si concentra il 21 CFR parte 11.
La firma elettronica deve essere unica per ciascun individuo. Prima dell’assegnazione della firma, l’identità dell’assegnatario deve essere verificata (§11.100).
Le firme manoscritte ed elettroniche devono essere legate ai rispettivi record in modo da non poter essere staccate, copiate o trasferite per falsificare un record elettronico (§11.70).
I record elettronici firmati devono contenere le seguenti informazioni legate alla firma:
- il nome in chiaro di chi firma
- la data e l’ora della firma
- il significato della firma (verifica, approvazione, presa visione, ecc. ) (§11.50 (a)).
La firma elettronica deve essere:
- composta di almeno due elementi distinti, ad esempio user-ID e password
- usata solamente dal suo proprietario
- amministrata in modo che il tentativo d’uso da parte di terzi richieda la collaborazione di due o più persone (§11.200 (a)).
Per quanto riguarda ID e password:
- devono essere unici
- la password deve avere una scadenza
- devono esserci meccanismi che rilevino e segnalino ripetuti tentativi di accesso falliti (§11.30).
Nel caso di utilizzo di un token:
- devono esserci procedure per disabilitare il token non appena si sospetta che sia stato perso, rubato o clonato;
- devono essere effettuati test iniziali e periodici dei token per assicurarne il corretto funzionamento.
Quindi se il software è usato per la produzione o il sistema qualità di un medical device è necessaria la convalida del sistema per assicurare che il sistema soddisfi il suo intended use.
Se il sistema memorizza dati elettronici serve anche la conformità al 21CFR part 11.
Articolo a cura di Giulia Colombo, Communication & Events Specialist di Quality Systems
Nella parte 2 del nostro approfondimento prenderemo in esame le linee guida per la convalida:
- ISO/TR 80002-2:2017 Medical device software – Part 2: Validation of software for medical device quality systems
- FDA General Principles of Software Validation; Final Guidance for Industry and FDA Staff, 2002
Scopri i nostri servizi di convalida per i Medical Device
Leggi altre GMP NEWS di Quality Systems


